Trisubstituted Aryl Cyclohexanecarboxylates (TACC): A Simple, New Molecular Scaffold for Antibiotics Design ()

1. Introduction
A tremendous increase in the worldwide spread of antibiotic-resistant bacteria has spurred a lot of interests in the search for antibiotics with new modes of action. Current drug discovery and development efforts are focused on modifying existing classes of antibacterial agents to improve potency and efficacy, provide broader spectrum of activity, reduce resistance and improve pharmacodynamics properties [1] . Others focus on identifying and screening compounds (natural or synthetic) that could act as inhibitors against unexploited, genomic targets [1] [2] . Biologically active molecules with novel chemical structures, acting against previously unexploited bacterial targets are more likely to be less prone to the existing compound- or target-based resistance mechanisms observed in most multi-drug resistant (MDR) strains of bacteria [3] . In fact, cellular pathways that are paramount to the survival of the bacterium at the early stages of the infection process have been identified as attractive candidates for rational drug design [4] . In these endeavors, Clp protease, which is one of the major cellular proteases responsible for degrading misfolded or damaged proteins and thus plays an essential role in maintaining protein function, has been established as a suitable target for new antibiotics [4] - [11] . Clp protease clade is an energy-dependent protease comprising of ATPases connected with diverse cellular activities (AAA+), like ClpX or ClpA in E. coli, or ClpX, ClpC, or ClpE in B. subtilis, and the subunit ClpP [8] [12] [13] . In Clp protease complex, the ATPase (ClpC and ClpX) is the regulatory subunit, while the ClpP subunit is the central proteolytic core [4] [8] [14] . Clp protease is an essential factor in controlling protein homeostasis and developmental processes like cell motility, genetic competence, cell differentiation, and sporulation [14] [15] [16] . Therefore, perturbation of the Clp protease complex could lead to severe physiological defects in bacteria, potentially leading to the bacterial demise [8] [17] . Proteolytic subunit of Clp protease, ClpP, was first identified in E. coli by Maurizi et al. [18] [19] and since then hundreds of studies have been done to understand its structure and mechanism of operation. The investigations of the crystal structures of ClpP from different species, including bacterial, human, plants and yeast [20] , have revealed that the protease is highly conserved [9] [21] [22] [23] . These crystal structures show that ClpP assembles into a tetradecameric barrel-shaped enzyme having an enclosed chamber that contains 14 serine proteolytic active sites [22] . Access to this ClpP proteolytic chamber is only possible through the two axial pores that are gated by the N-terminal region of the protomers [22] [24] . Although the proteolytic chamber is large enough to accommodate a 50 kDa protein, the tapered axial pores prevent the entry of even the smallest folded protein [23] . So ClpP protease depends on its partners, the highly specific AAA+ proteases, to recognize native proteins, unfold them and spool the denatured polypeptide into the proteolytic chamber for degradation [21] . The importance of ClpP protease in intracellular milieu has made it an attractive target for new antibiotics. Its inhibition by cyclic peptides [25] , β-lactones [17] [26] [27] [28] [29] , and its activation by acyldepsipeptides (ADEPs) [5] [6] [12] [30] are detrimental to different bacterial strains.
In our continued studies of bactericidal agents and ClpP activation/deactivation [5] [17] [30] , we came across new classes of compounds called Activator of Cylindrical Proteases (ACP) reported by Leung et al. [31] . These were four different structural classes of compounds with no structural similarities to previously reported ADEPs [4] , but with comparable bactericidal activities against different pathogens [31] . Leung and co-worker attributed antibacterial activities of these ACPs to CpP activation suggesting similarity in mechanism of actions of ADEPs and ACPs. They proposed that ACPs prevent ClpP from binding to its associated unfoldase, while concurrently promoting nonspecific proteolysis probably via the opening of the axial pores. They also proposed the existence of an additional pocket, the C pocket [31] , in conjunction with the previously reported H pocket [4] , that helps enhance compound binding. Of the four structural classes reported, our attention was drawn to ACP 5 and 4 (ACP 4 has p-nitro in place of p-bromo) (Scheme 1) since they were considered unsuitable for further structural optimization because of the challenges access to the structure poses in a structure activity relationship (SAR) studies [31] , even though they showed significant antibacterial activities. We herein report the syntheses, structure activity relationship and antibacterial activities against B. mycoides and B. subtilis of ACP 5 and 4 and their structural analogues. We also present, herein, evidences that suggest that there is possibly a synergistic mechanism of action of these new class of compounds involving membrane permeabilization and a minimal amount (if at all present) of ClpP activation. Since the core structure of these compounds is a cyclohexane carboxylate, we have chosen to tag this new antibiotic scaffold a trisubstituted aryl cyclohexane carboxylate (TACC). We varied the substituents on both the aryl group and the cyclohexane ring in our SAR studies, and the antibacterial activity results of the different analogues thus obtained are herein presented. To the best of our knowledge, synthesis and medicinal application of these TACCs have not been reported in the literature before now.
2. Results and Discussion
2.1. Synthesis of Dichlorovinyl TACCs 20-29 and Preliminary Antibacterial Activity Studies on Bacillus mycoides
Our initial synthetic target was ACP 5 (Scheme 1). The goal was to find a simple way to assemble the core structure in the minimal possible steps to facilitate diversity-oriented synthesis of analogues for SAR. In our proposed retrosynthesis (Scheme 1), the core structure could be obtained by a pseudo-Robinson annulation reaction (tandem Michael-Aldol addition reaction) of the conjugated 2,4-dienone 2 with ethyl acetoacetate 3. The conjugated ketone 2 could then be synthesized from cross Aldol condensation of dichloroacrolein 4 and p-bromoacetophenone. The synthetic challenge here was making dichloroacrolein 4 which was not commercially available.
With little modifications of a previously reported procedure [32] , dichloroacrolein 4 was synthesized on multi-gram scale in good yield by radical reaction of isobutyl viny ether with carbon tetrachloride using benzoyl peroxide as radical initiator (Scheme 2). The reagents were simple, but the process was elaborate because of the propensity of 4 to easily polymerize (black polymeric tar was seen in some cases). The presence of both intermediates 1,3,3,3-tetrachloropropyl isobutyl ether 6 and 1,3,3-trichloro-2-propenyl isobutyl ether 7 was confirmed by quick proton NMR of an aliquot of the reaction mixture. With dichloroacrolein 4 in hand, its cross-Aldol condensation was conducted with different aryl ketones taken into account the potential electronic effect of the aryl substituents on the alcohol functional group of the desired TACCs (Table 1). The chalcones 10-19 thus obtained were then reacted with ethyl acetoacetate 3 in a tandem
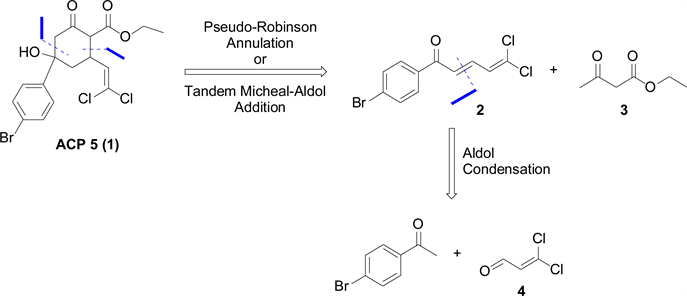
Scheme 1. ACP 5 (1) and its proposed retrosynthesis.
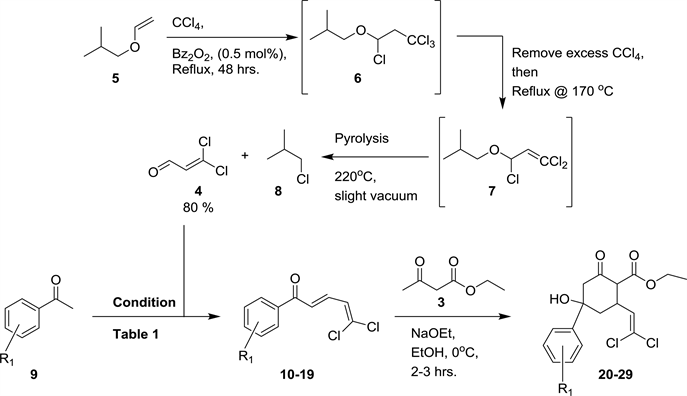
Scheme 2. Synthetic route to 3,3-dicholoroacrolein and synthesis of TACC 20-29.
![]()
![]()
Table 1. Synthesis of trisubstituted aryl cyclohexanecarboxylate (TACC) 20-29.
Michael-Aldol addition reaction to afford the corresponding TACCs 20-29 (Scheme 2). The conditions and yields for the different reactions are presented in Table 1.
Our initial antibacterial inhibitory activities studies of TACCs 20-29 was done on Bacillus mycoides (Figure 1, Table 2, entries 1-10). All our synthesized dichlorovinyl compounds 20-29 showed promising inhibitory activities. It was interesting to note that TACC 20 (ACP 5, Table 2, entry 1, MIC = 25 µg/mL) was more active than TACC 25 (ACP 4, Table 2, entry 6, MIC = 100 µg/mL), which is in contrast to what was reported by Leung et al. [31] .
The same trend was observed for these two compounds in our later antibacterial activities studies against B. subtilis (Table 3, entry 1: MIC = 100 µg/mL for ACP 5; entry 6: MIC > 200 µg/mL for ACP 4). The most active of the dichlorovinyl analogues was TACC 22 (MIC = 15 µg/mL). However, this compound was very unstable at room temperature and its decomposition was observed in a closed vial after some hours of storage.
This probably explained the dark coloration seen when a solution of 22 in DMSO was prepared for bioassay analysis. It was unclear whether the compound itself or its degradation product was responsible for the increased antibacterial activity. So, to avoid any ambiguity in bioassay data analysis, a very stable p-bromo analogue 20 (MIC = 25 µ/mL) was chosen for further structural modification for SAR. Bearing in mind the need for a potential drug-lead to have acceptable ADME/Toxicity properties and ability to overcome various barriers in living systems [33] , we concur with Leung et al. that ACP 5 (TACC 20) do not have drug-like structure possibly because of the liability the dichlorovinyl moiety poses. Establishing the pharmacophoric groups in this new antibiotic molecular scaffold is of great importance to us as this would aid us in identifying the group we could play within chemical proteomics studies without jeopardizing the antibacterial activity of the molecule. So, we decided to sequentially analyze the effect of each functional group in the molecule on its antibacterial activity. We started off by substituting the dichlorovinyl handle with other groups ensuring that the hybridization of the connecting carbon to the cyclohexane carboxylate
![]()
Figure 1. Structures of trisubstituted aryl cyclohexane carboxylate (TACC) tested against B. mycoides and B. subtilis.
![]()
Table 2. Antibacterial activities of trisubstituted aryl cyclohexane carboxylate (TACC) against B. mycoides.
(a) *ACP 5; **ACP 4. (b) Minimum Inhibitory concentration (MIC) was defined as the lowest compound concentration able to completely inhibit bacterial growth for up to 48 hours. (c) ACP 5, and TACCs 22, 27, showed activity up to 72 hours.
ring is maintained.
2.2. Evaluation of the Effect of gem Dichlorovinyl Substituent on TACCs’ Antibacterial Activities: Synthesis of TACCs 40-44 and Their Antibacterial Activities against B. mycoides
To study the effect of the dichlorovinyl handle on the activity of TACC 20, the dichlorovinyl moiety was replaced by dimethylvinyl 40, thiophenyl 41, furanyl 42, 5-methylfuranyl 43 and phenyl 44 substituents (Table 4, entries 1-5). The idea was to study the role of the electrophilic character of the dichlorovinyl
![]()
Table 3. Antibacterial activities of trisubstituted aryl cyclohexane carboxylate (TACC) against B. subtilis.
(a) *ACP 5; **ACP 4. (b) Minimum Inhibitory concentration (MIC) was defined as the lowest compound concentration able to completely inhibit bacterial growth for up to 48 hours. (c) ACP 5, and TACCs 22, 27, showed activity up to 72 hours.
substituent on the antibacterial activity of the whole molecule since the vinyl gem dihalide functionality is known to be versatile bidentate electrophile [34] . Thus TACCs 40-44 were synthesized by cross Aldol condensation of 4’-bromoacetophenone with the appropriate aldehydes under basic condition to generate the corresponding chalcones 31-35 which were then annulated via tandem Michael-Aldol reaction (Table 4, entries 1-5). These compounds were tested against B. mycoides and the antibacterial activity data are presented in Table 2, entries 11-15. TACC 42 (Table 2, entry 13, MIC = 50 µ/mL) was the most active of the five analogues, whereas TACC 40 (Table 2, entry 11, MIC > 200 µ/mL) was not active at all. Compound 43 (Table 2, entry 14, MIC > 50 µ/mL) has slightly reduced activity than TACC 42. These observations from 40, 42, and 43, interestingly point to some sort of synergistic contribution of the electrophilic nature of this side handle to the antibacterial activity of the molecule. Although the introduction of additional oxygen atom into the molecule by the furanyl moiety increases the nucleophilic character and hydrogen-accepting ability of the side handle, the decrease in activity observed for compound 43 suggests that electrophilicity of the side handle may have more role to play in the antibacterial activity of the molecule, more so that the dichlorovinyl moiety has more electrophilic character and was observed to be more active. We were excited to notice though, that TACC 42 has comparable antibacterial activity with ACP 5. Thus TACC 42 was chosen for further structural optimization to improve potency.
2.3. Evaluation of the Effect of the Hydroxyl and the Oxo-(Ketone) Functional Groups on TACCs’ Antibacterial Activities: Synthesis of TACCs 45-52 and Their Antibacterial Activities against B. mycoides
The tertiary hydroxyl functional group on TACCs is a potential hydrogen donating
and hydrogen accepting group. The nature of the aryl group connected to this tertiary carbon center can affect these properties. Compounds 45-48 (Table 4, entries 6-9) were synthesized to evaluate this effect. With TACC 42 and the result of its activity in hand, compound 45 was synthesized to compare the effect of the position of the bromo group on the activity of the molecule. It turns out that 42 (Table 2, entry 13, MIC = 50 µ/mL) and 45 (Table 2, entry 13, MIC = 50 µ/mL), with para- and meta-bromophenyl substituents respectively, have the same activities against B. Mycoides. The same trend was also observed for TACCs 20 and 21 (Table 2, entries 1 and 2 respectively). Introduction of a very strong electron-withdrawing group on the benzene ring, nitro group in 47, and an electron-donating group, methyl group in 48, both seem to lead to loss of activity in the molecule (Table 2, entries 18 and 19, MIC in both cases > 200).
A chemoselective hydride reduction of the oxo-(ketone) group in compound 22 yielded a more potent analogue 50 with MIC of 8 µg/mL against B. Mycoides (Table 2, entry 20). Also compound 54, synthesized using acetone as the Michael donor instead of the ethyl acetoacetate, had a more pronounced activity against B. Mycoides (Table 2, entry 22, MIC = 15 µg/mL) than any of its furanyl counterparts with the carboxylate group.
2.4. Antibacterial Activity Study of Selected TACCs against B. subtilis
Some library of our synthesized TACCs (Figure 1) were tested against B. subtilis for antibacterial activity.
The results for these analyses are presented in Table 3. It is interesting to point out that most of the trends recorded for these compounds’ activities against B. Mycoides (Table 2) were observed here as well: TACC 20 (ACP 5) was more active than TACC 25 (ACP 4) (Table 3, entries 1 and 6 respectively), which is in contrast to what was reported by Leung et al. [31] ; Antibacterial activity decreases as you go from electron-deprived aryl substituents to electron-rich aryl group at the quaternary carbon center with the tertiary alcohol (Table 3, entries 1, 3 and 10); Substituting the chemically liable, commercially unavailable dicholorovinyl moiety, which possesses poor drug-like property [31] with a commercially available and stable furanyl analogue resulted in a comparably active compound (Table 3, entries 1 and 12 respectively); A 2-hydroxycyclohexane carboxylate proved to be more active than the corresponding 2-oxocyclohexane carboxylate (Table 3, entries 15-16, and entries 1 and 3 respectively).
An interesting observation was made while evaluating TACCs’ antibacterial activity. Activity for these compounds seemed to diminish over time. Minimum inhibitory concentrations (MIC) were determined by the agar dilution method [35] . When the agar plates were inspected at 24, 48, and 72 hours, the bacterial growth tended to steadily increase over time. In typical agar dilution MIC assays, there is little change in bacterial growth from 24 to 48 hours and no change from 48 to 72 hours. The peculiar activity of these TACCs is indicative of gradual compound degradation in the growth medium. As the concentration of active compound decreases over time, persistent bacterial cells are eventually able to proliferate.
We reasoned that the loss in activity over time could be a result of compound dehydration. Dehydration could occur either by a base promoted E1cB mechanism or an acid promoted E1 mechanism (Figure 2(a)). In the growth media, both mechanisms could be operative. To test the effect of dehydration on TACC activity, compound 55, which was recovered as a byproduct from syntheses of 22, was tested against B. subtilis and found to be completely inactive. Apparently, the tertiary alcohol is absolutely essential for antibacterial activity.
The report by Leung and co-workers [31] suggests that antibacterial activity of TACC 25 (ACP 4) and TACC 20 (ACP 5) was due to activation of the peptolytic activity of ClpP. To confirm this mechanism of action, we tested TACC 20 (ACP 5) and compound 22 against a ∆clpP-spx null strain of B. subtilis that is not susceptible to the ADEPs [30] [36] [37] .
To our surprise, TACCs were more active against the B. subtilis ∆spx null strain and ∆clpP-spx double null strain than the B. subtilis wild type strain (Figure 3(a)). These data suggest that TACCs have targets other than ClpP. We also tested TACCs for their ability to activate ClpP in vitro. We found that TACCs mediated very weak ClpP activation compared to ADEP1 (Figure 3(b)). At the highest concentration tested (1000 µM), TACC 20-induced decapeptide hydrolysis was only slightly more than in blank samples with no activator. ADEP1 on the other hand appears to saturate ClpP at 1000 µM.
Our experimental results points to the fact that these new antibiotic molecular scaffolds have target other than ClpP that is responsible for antibacterial activity. In fact, preliminary bacterial cytological profiling (BCP) studies on these new class of antibiotics indicate that they are membrane active compounds and disrupt membrane integrity through rapid membrane permeabilization. Therefore, the mode of action of these compounds could be a synergistic one involving a very minimal ClpP activation and a pronounced membrane permeabilization.
![]()
Figure 2. Effects of TACC dehydration and reduction on antibacterial activity: (a) Possible mechanisms of TACC dehydration and effect on antibacterial activity; (b) Changes of antibacterial activity with time.
![]()
Figure 3. Attempts to Validate ClpP as TACC target: (a) Antibacterial activity against B. subtilis, wild type, spx null strain and ClpP/spx double null strain; (b) Hydrolysis of a fluorogenic decapeptide substrate (15 μM) by E. coli ClpP (25 nM) was assayed in the presence of increasing concentrations of ADEP1 or TACC 20 (ACP5). Bars represent initial rates of decapeptide hydrolysis (average of 2 experiments). Error bars indicate standard deviation. Horizontal red dashed line indicates decapeptide hydrolysis rate in the absence of ADEP.
3. Conclusion
A new class of antibacterial agents has been synthesized on a new molecular scaffold of cyclohexane carboxylate. We have tagged these new compounds TACCs (Trisubstituted Aryl Cyclohexane Carboxylate). These new molecules are structural analogues of ACP 4 and 5 previously reported by Leung et al. [31] , and were synthesized to circumvent the drug-like property (drug-ability) challenges and liability noted in ACP 4 and 5. The TACCs exhibited equal, and in some cases better, antibacterial activity than ACP 4 and 5 (Table 2 and Table 4). The tertiary alcohol on the quaternary carbon center of the cyclohexane carboxylate was found to be crucial to the antibacterial activity of this class of compounds. It was also discovered through the extensive bioassay analyses conducted that the 2-hydroxycyclohexane carboxylate (hydroxyl-TACC) was more active than the corresponding 2-oxocyclohexane carboxylate (oxo-TACC). While ClpP activation in TACCs is very weak, the preliminary bacterial cytological profiling (BCP) study revealed that this class of compounds exhibits pronounced membrane permeabilization leading to disruption of bacterial membrane integrity.
4. Experimental
4.1. General
All chemicals were purchased from Sigma-Aldrich and used without further purification. NMR analyses were conducted on Bruker Avance Ultrashield Spectrometer in d-DMSO solvent (400 MHz or 600 MHz for 1H and 100 MHz for 13C NMR). Residual DMSO signal was used as an internal reference (2.52 ppm for 1H and 40 ppm for 13C).
4.2. Synthesis
4.2.1. Synthesis of 3,3-Dichloroacrolein (4)
To a solution of carbon tetrachloride (200 mL, 317.34 g, 2.06 mol, 6.8 eq.) and isobutyl vinyl ether (39.5 mL, 30.35 g, 0.30 mol, 1 eq.) in a 1L 2-neck round bottom flask, equipped with a magnetic stirring bar was added catalytic amount of benzoyl peroxide (0.60 g, 2.50 × 10−3 mol, 8.2 × 10−3 eq.). The reaction mixture was then refluxed for 48 hr. Upon cooling, the refluxing setup was replaced with a fractional distillation setup, excess carbon tetrachloride was removed by distillation and the residual liquid was heated to 170˚ - 196˚ where evolution of large amount of HCl was observed. The residue was then slowly heated to 220˚ under slight vacuum and different fractions were collected. 3,3-dichloroacrolein was obtained in 80% pure yield after redistillation using a short path, bp 124˚ - 126˚ (atm). 1H NMR (400 MHz, CDCl3) δ (ppm) 6.45 (d, 1 H, J = 6.8 Hz), 9.88 (d, 1 H, J = 6.8 Hz).
4.2.2. Synthesis of TACC 20-Ethyl 4-(4-Bromophenyl)-2-(2,2-dichloroethenyl)-4-hydroxy -6-oxocyclohexanecarboxylate
To a solution of 1.5 eq. of NaOH in 4 mL of water was added solution of 4’-bromoacetophenone (6.03 mmol, 1.0 eq.) in ethanol (6 mL). The mixture was stirred for 5 - 10 minutes and then a solution of 3,3-dicholroacrolein (6.63 mmol, 1.1 eq.) in 2 mL of ethanol was slowly added. Solid chalcone product 10 started forming almost instantaneously. Chalcone 10 was filtered after 20 minutes, washed with cold ethanol and dried. To a separate solution of sodium ethoxide (21% NaOEt in EtOH, 1.3 eq.) in ethanol (3 mL) was added ethyl acetoacetate (0.72 mmol, 1.1 eq.). The mixture was stirred for 10 minutes followed by the addition of chalcone 10 (0.65 mmol, 1.0 eq.). Then the reaction was left to stir for 5 hr. Upon completion of the reaction as monitored by TLC, ethanol was evaporated in vacuo and the reaction mixture was poured into water, extracted with ethyl acetate (3 × 15 mL), washed with brine and dried over anhydrous sodium sulfate. The drying agent was filtered off and the organic solvent was evaporated in vacuo to afford TACC 20 in 95% yield as a thick reddish oil. 1H NMR (400 MHz, DMSO-d6), δ (ppm) 1.21 (t, 3H, J = 7.1 Hz, CH3), 1.76 (d, 1H, J = 12.5 Hz, CH), 2.24 (t, 1H, J = 12.6 Hz, CH), 2.35 (d, 1H, J = 1.8 Hz, CH), 3.14 (d, 1H, J = 13.8 Hz, CH), 3.62 (m, 1H, CH), 3.77 (d, 1H, J = 11.8 Hz, CH), 4.15 (m, 2H, CH2), 5.82 (s, 1H, CH), 6.10 (d, 1H, J = 9.8 Hz, CH), 7.44 (d, 2H, J = 8.5 Hz), 7.57 (d, 2H, J = 8.5 Hz). MS (ESI, m/z) 437 [M + H]+ for C17H17BrCl2O4.
4.2.3. General Procedure for the Synthesis of TACCs 21-29
To a mixture of substituted acetophenone (7.2 mmol, 0.9 eq.) and 3,3-dicholroacrolein (8.0 mmol, 1.0 eq.) in acetic acid (10 mL) was added 1.2 mL of H2SO4. After 24 hours, the reaction mixture was poured on ice (with some water) and the precipitate that formed was filtered and washed with ice-cold water to afford chalcones 11-19 (Note: Chalcone 16 did not yield a solid precipitate at this point, thus its reaction mixture was extracted with dichloromethane (25 mL × 3), the obtained organic layer was washed with water and brine and then dried over anhydrous sodium sulfate. The drying agent was filtered, and the organic solution was evaporated in vacuo to furnish chlcone 16). To a separate solution of sodium ethoxide (21% NaOEt in EtOH, 2.0 eq.) in ethanol (10 mL) was added ethyl acetoacetate (5.15 mmol, 2.0 eq.). The mixture was stirred for 10 minutes followed by the addition of the appropriate chalcone (2.57 mmol, 1.0 eq.). Then the reaction was left to stir for 2 - 3 hr. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured on iced-water and then acidified using 1 M HCl. Solid precipitate was filtered from the mixture and washed with ice-cold water to obtain crude TACCs 21-29. These were then purified using flash column chromatography (Hexane:Ethyl acetate/4:1).
1) TACC 21—Ethyl 4-(3-Bromophenyl)-2-(2,2-dichloroethenyl)-4-hydroxy- 6-oxo cyclohexanecarboxylate
Isolated as yellow solid (97%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.21 (t, 3H, J = 4.7 Hz), 1.75 (d, 1H, J = 6.3 Hz), 2.26 (t, 1H, J = 8.4 Hz), 2.36 (d, 1H, J = 1.6 Hz), 3.17 (d, 1H, J = 9.2 Hz), 3.60 (dd, 1H, J = 9.2 Hz), 3.77 (d, 1H, J = 7.9 Hz), 4.15 (m, 2H), 5.87 (s, 1H), 6.09 (d, 1H, J = 6.6 Hz), 7.34 (t, 1H, J = 5.2 Hz), 7.47 (d, 1H, J = 8.5 Hz), 7.49 (d, 1H, J = 5.2 Hz), 7.68 (s, 1H). 13C NMR, δ (ppm) 14.6, 39.1, 41.9, 53.7, 61.3, 61.4, 76.4, 122.1, 123.0, 124.5, 128.7, 131.1, 131.3, 132.1, 150.9, 169.2, 202.5. MS (ESI, m/z) 437 [M + H]+, 459 [M + Na]+ for C17H17BrCl2O4.
2) TACC 22—Ethyl 4-(4-Chlorophenyl)-2-(2,2-dichloroethenyl)-4- hydroxy-6-oxo cyclohexanecarboxylate
Isolated as light yellow solid (90%) 1H NMR (600MHz,DMSO-d6) δ (ppm) 1.20 (t, 3 H, J = 7.2 Hz), 1.81 - 1.68 (m, 1 H), 2.22 (t, 1 H, J = 12.7 Hz), 2.36 (dd, 1 H, J = 2.2, 13.6 Hz), 3.13 (d, 1 H, J = 13.6 Hz), 3.61 (dt, 1 H, J = 9.9 Hz), 3.76 (d, 1 H, J = 11.7 Hz), 4.21 - 4.04 (m, 2 H), 5.81 (s, 1 H), 6.09 (d, 1 H, J = 9.9 Hz), 7.45 - 7.36 (m, 2 H, J = 8.8 Hz), 7.57 - 7.45 (m, 2 H, J = 8.8 Hz). 13C NMR (DMSO-d6) δ (ppm) 14.1, 37.9, 40.6, 52.5, 59.8, 60.4, 74.9, 120.0, 126.6, 128.0, 131.4, 131.6, 146.3, 168.5, 203.0. HRMS (ESI) Predicted for [C17H17Cl3O4 + H]+: 391.0271, found 391.0260
3) TACC 23—Ethyl 4-(2-Chlorophenyl)-2-(2,2-dichloroethenyl)-4-hydroxy- 6-oxo cyclohexanecarboxylate
Isolated as bright yellow solid (85%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.22 (t, 3H, J = 6.9 Hz), 1.75 (d, 1H, J = 6.4 Hz), 2.1 (s, 1H), 2.2 (d, 1H, J = 9.5 Hz), 2.41 (d, 1H, J = 12 Hz), 2.74 (t, 1H, J = 12.8 Hz), 3.38 (d, 1H, J = 14.2 Hz), 3.61 (m, 1H), 3.82 (d, 1H, J = 11.7 Hz), 4.15 (q, 2H, J = 7.2 Hz), 6.02 (s, 1H), 6.21 (d, 1H, J = 9.6 Hz), 7.33 (m, 2H), 7.40 (d, 1H, J = 7 Hz), 7.72 (d, 1H, J = 7.7 Hz). 13C NMR, δ (ppm) 14.5, 37.4, 38.1, 39.5, 50.2, 60.7, 60.8, 75.7, 120.4, 127.8, 128.3, 129.7, 131, 131.8, 143, 169, 203. MS (ESI, m/z) 391 [M + H]+, 413 [M + Na]+ for C17H17Cl3O4
3) TACC 24—Ethyl 4-(3-Chlorophenyl)-2-(2,2-dichloroethenyl)-4- hydroxy-6-oxo cyclohexanecarboxylate
Isolated as yellow solid (95%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.2 (t, 3H, J = 7 Hz), 1.73 (d, 1H, J = 12.6 Hz), 2.26 (d, 1H, J = 12.9 Hz), 2.34 (d, 1H, J = 1.6 Hz), 3.16 (d, 1H, J = 13.7 Hz), 3.6 (m, 1H), 3.76 (d, 1H, J = 11.8 Hz), 4.14 (m, 2H), 5.86 (s, 1H), 6.09 (d, 1H, J = 9.8 Hz), 7.4 (m, 3H), 7.52 (s, 1H). 13C NMR, δ (ppm) 14.5, 38.4, 41, 52.9, 60.3, 60.9, 75.4, 120.5, 123.8, 125.2, 127.4, 130.6, 131.8, 133.5, 150.4, 169, 203. MS (ESI, m/z) 391 [M + H]+ for C17H17Cl3O4
4) TACC 25—Ethyl 2-(2,2-dichloroethenyl)-4-hydroxy-4-(4-nitrophenyl)- 6-oxo cyclohexanecarboxylate
Isolated as a yellow solid (73%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.22 (t, 3H), 1.81 (d, 1H), 2.41 (m, 2H), 2.52 (s, 1H), 3.24 (d, 1H, J = 13.6 Hz), 3.67 (m, 1H), 3.83 (d, 1H, J = 11.6 Hz), 4.16 (m, 2H), 6.1 (d, 2H, J = 6.4 Hz), 7.78 (d, 2H, J = 7.8 Hz), 8.25 (d, 2H, J = 7.9 Hz). 13C NMR, δ (ppm) 14.5, 38.4, 40.2, 52.6, 60.3, 60.9, 75.7, 120.7, 123.8, 126.6, 131.7, 146.9, 155, 168.9, 203. MS (ESI, m/z) 402 [M + H]+, 424 [M + Na]+ for C17H17Cl2NO6.
7) TACC 28—Ethyl 2-(2,2-dichloroethenyl)-4-hydroxy-6-oxo-4-phe- nylcyclohexane carboxylate
Isolated as bright yellow solid (95%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.22 (t, 3H), 1.78 (d, 1H, J = 2.2 Hz), 2.27 (d, 1H, J = 12.8 Hz), 2.4 (d, 1H, J = 11.9 Hz), 3.15 (d, 1H, J = 13.7 Hz), 3.65 (m, 1H), 3.79 (d, 1H, J = 11.8 Hz), 4.12 (m, 2H), 6.12 (d, 1H, J = 9.8 Hz), 7.37 (t, 1H, J = 7.4 Hz), 7.47 (t, 2H, J = 5.1 Hz), 7.52 (d, 2H, J = 9.8 Hz). 13C NMR, δ (ppm) 14.5, 39.8, 40.2, 53.2, 60.3, 60.8, 75.6, 120, 125, 127.4, 128.6, 131.9, 147.8, 169.1, 203.8. MS (ESI, m/z) 357 [M + H]+, 379 [M + Na]+ for C17H17Cl2NO6.
8) TACC 29—Ethyl 2-(2,2-dichloroethenyl)-4-hydroxy-4-(4-methylphenyl)- 6-oxo cyclohexanecarboxylate
Isolated as a yellow solid (97%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.21 (t, 3H, J = 7 Hz), 1.75 (d, 1H, J = 12.4 Hz), 2.22 (t, 1H, J = 11.6 Hz), 2.29 (s, 3H), 2.36 (d, 1H, J = 12 Hz), 3.1 (d, 1H, J = 13.7 Hz), 3.62 (m, 1H), 3.77 (d, 1H, J = 11.8 Hz), 4.15 (q, 2H, J = 7.2 Hz), 5.6 (s, 1H), 6.11 (d, 1H, J = 9.8 Hz), 7.17 (d, 2H, J = 8 Hz), 7.36 (d, 2H, J = 8 Hz). 13C NMR, δ (ppm) 14.5, 21, 38.4, 39.5, 41.3, 53.3, 60.3, 60.8, 75.5, 79.3, 120.3, 124.9, 129.1, 132, 136.4, 144.9, 169.1, 203.8. MS (ESI, m/z) 371 [M + H]+, 393 [M + Na]+ for C18H20Cl2O4.
4.2.4. General Procedure for the Synthesis of TACCs 40-48
To a solution of substituted acetophenone (6.06 mmol, 1.0 eq.) and aldehyde (9.08 mmol, 1.5 eq.) in methanol (20 mL) was added 3 M NaOH (2 mL) dropwise. Solid precipitate crashed out of solution and was filtered, washed with cold methanol and dried in vacuo to afford corresponding chalcones 31-39. The chalcones were used without further purification. To ethanol (10 mL) in a round bottom flask equipped with a stirring bar was added sodium ethoxide (21% NaOEt in ethanol, 4.88 mmol, 2.0 eq.) and ethyl acetoacetate (4.88 mmol, 2.0 eq.) respectively. The solution was stirred at room temperature for 15 minutes, cooled to 0˚C and then stirred for additional 15 minutes. Chalcone was slowly added to the solution and the reaction mixture was allowed to warm up to room temperature. Stirring continued at room temperature until the reaction was complete as evident on TLC. Some ethanol was evaporated in vacuo from the reaction mixture and the resulting solution was poured on iced water. The solution was extracted with ethyl acetate and the extract was washed with brine, dried over anhydrous sodium sulfate and the drying agent was filtered to obtain the crude product. The crude product was purified using column chromatography (EtOAc:Hexane/1:3) to afford pure TACCs 41, 42, 45 and 46.
1) TACC 41—Ethyl 4-(4-Bromophenyl)-4-hydroxy-6-oxo-2-(2-thiophenyl)- cyclohexane carboxylate
Isolated as yellow solid (68%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.09 (t, 3H, J = 7.1 Hz), 2.05 (m, 1H), 2.38 (d, 1H, J = 11.5 Hz), 2.5, (m, 2H), 3.28 (d, 1H, J = 13.8 Hz), 3.38 (m, 1H), 4.04 (q, 2H, J = 7.3 Hz), 5.83 (s, 1H), 6.98 (d, 2H, J = 3.5 Hz), 7.37 (t, 1H, J = 3.3 Hz), 7.48 (d, 2H, J = 6.8 Hz), 7.56 (d, 2H, J = 4.2 Hz). 13C NMR, δ (ppm) 14, 38, 41, 46, 53, 61, 64, 75, 121, 124, 127, 132, 146, 147, 148, 169, 204. MS (ESI, m/z) 407 for C19H17BrO3S.
2) TACC 42—Ethyl 4-(4-Bromophenyl)-2-(2-furanyl)-4-hydroxy-6- oxocyclohexane carboxylate
Isolated as yellow solid (75%). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 1.13 (t, 3H, J = 7.1 Hz), 2.05 (m, 1H), 2.37 (m, 2H), 3.26 (d, 1H, J = 13.7 Hz), 4 (m, 2H), 4.08 (q, 2H, J = 4.4 Hz), 5.8 (s, 1H), 6.14 (d, 1H, J = 3.2 Hz), 6.38 (s, 1H), 7.48 (d, 2H, J = 8.6 Hz), 7.57 (d, 3H, J = 8.3 Hz). 13C NMR, δ (ppm) 14.5, 36, 42.2, 53, 60.3, 60.7, 75, 105.6, 110.7, 120.6, 127.5, 131.4, 142.4, 147.4, 156, 169.3, 204. MS (ESI, m/z) 391 [M + H]+ for C19H17BrO4.
3) TACC 45—Ethyl 4-(3-Bromophenyl)-2-(2-furanyl)-4-hydroxy-6- oxocyclohexane carboxylate
Isolated as yellow solid (65%). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 1.14 (t, 3H, J = 7.1 Hz), 1.99 (s, 1H), 2.48 (d, 1H, J = 1.8 Hz), 2.50 (d, 1H, J = 1.8 Hz), 2.51 (s, 1H), 3.26 (d, 1H, J = 13.7 Hz), 3.9 (m, 2H), 4.08 ( m, 2H), 5.86 (s, 1H), 6.16 (s, 1H), 6.37, (s, 1H), 7.45 (t, 1H, J = 1.1 Hz), 7.46 (d, 1H, J = 5.9 Hz), 7.47 (d, 1H), 7.52 (s, 1H). 13C NMR, δ (ppm) 14.5, 36, 42.2, 52.9, 60.3, 60.8, 75.1, 105.7, 110.9, 122.2, 124.2, 127.9, 130.2, 130.9, 142.3, 150.5, 155.9, 169.4, 204.2.
4) TACC 46—Ethyl 4-(4-Chlorophenyl)-2-(2-furanyl)-4-hydroxy-6- oxocyclohexane carboxylate
Isolated as yellow solid (65%). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 1.14 (t, 3H, J = 7.1 Hz), 2.06 (m, 1H), 2.41 (m, 2H), 3.26 (d, 1H, J = 13.8 Hz), 3.96 (m, 2H), 4.08 (q, 2H, J = 2.8 Hz), 5.83 (s, 1H), 6.15 (d, 1H, J = 3.2 Hz), 6.38 (d, 1H, J = 2.8 Hz), 7.43 (d, 2H, J = 8.6 Hz), 7.55 (d, 2H, J = 3.4 Hz). 13C NMR, δ (ppm) 14.5, 36, 42.2, 53.1, 60.3, 60.7, 75, 105.6, 110.9, 127.1, 128.5, 132, 142.4, 147, 156.1, 169.3, 204. MS (ESI, m/z) 363.1 [M + H]+, 385.1 [M + Na]+ for C19H19ClO5.
4.2.5. General Procedure for the Chemoselective Hydride Reduction of Oxo-TACCs—Synthesis of Hydroxy-TACCs 49-52
Corresponding oxo-TACC (1 mole eq.) was dissolved in methanol and treated with sodium borohydride (2 moles eq.). Upon completion, as determined by TLC, the solvent was removed in vacuo. The concentrated residue was diluted with ethyl acetate (~10 mL) and extracted once with saturated aqueous ammonium chloride. The aqueous extract was back extracted once with ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate. The desired product was isolated by flash chromatography on silica gel using a 30% ethyl acetate in hexanes mobile phase.
1) TACC 50—Ethyl 4-(4-Chlorophenyl)-2-(2,2-dichloroethenyl)-4,6- dihydroxy-6-oxo cyclohexanecarboxylate
Obtained as yellow solid (89%) 1H NMR (600MHz,DMSO-d6) δ (ppm) 1.19 (t, 3H, J = 7.2 Hz), 1.68 - 1.61 (m, 1 H), 1.81 (t, 1H, J = 12.5 Hz), 1.87 - 1.82 (m, 1 H), 1.99 (dd, 1H, J = 2.9, 14.3 Hz), 2.72 (dd, 1H, J = 2.8, 11.6 Hz), 3.41 - 3.36 (m, 1 H), 4.03 (dq, 1H, J = 7.0, 10.9 Hz), 4.12 (dq, 1H, J = 7.0, 10.8 Hz), 4.34 (q, 1H, J = 3.1, 6.5 Hz), 5.53 (d, 1H, J = 6.6 Hz), 5.87 (s, 1 H), 6.00 (d, 1H, J = 9.5 Hz), 7.38 (d, 2H, J = 8.8 Hz), 7.46 (d, 2H, J = 8.8 Hz). 13C NMR (DMSO-d6) δ (ppm) 14.1, 31.4, 41.5, 41.9, 50.5, 59.8, 68.3, 72.9, 118.2, 126.5, 127.9, 131.2, 133.8, 147.1, 171.6. HRMS (ESI) Predicted for [C17H19Cl3O4 + Na]+: 415.0247, found 415.0239.
Minimum inhibitory concentration determinations: B. subtilis MICs were determined using standard agar dilution techniques. Liquid cultures (LB Broth) inoculated from a fresh single colony were grown for 6 hours at 37˚C. LB agar plates supplemented with varying concentrations of test compound were inoculated with 5 µL of the liquid culture and then incubated at 37˚C for up to 72 hours. The agar plates were inspected for growth at 24-hour intervals. The MIC was determined to be lowest concentration of compound able to completely inhibit B. subtilis growth after 48 hours.
Acknowledgements
We are grateful for the financial support provided by Brown University.
We appreciate Prof. Joseph Pogliano’s laboratory of the University of California San Diego for conducting preliminary bacterial cytological profiling on some of these compounds.